---
title: "实验2:差异表达分析与可视化"
subtitle: "使用DESeq2识别差异基因并进行功能分析"
execute:
freeze: false
eval: true
echo: true
output: true
warning: false
message: false
---
## 实验目标
完成本实验后,你将能够:
1. 使用DESeq2进行差异表达分析
2. 理解DESeq2结果表的各列含义
3. 筛选和解释差异表达基因
4. 绘制火山图、热图等可视化图表
5. 进行基础的功能富集分析(GO/KEGG)
- **实验时长**:45分钟
- **实验数据**:真实甲状腺癌RNA-seq数据(PTC vs ATC)
- **数据下载**:<a href="../data/geneCountMatrix.txt" download>geneCountMatrix.txt</a> | <a href="../data/samplesinfo.txt" download>samplesinfo.txt</a>
---
## 关键知识点
::: {.callout-important}
### 核心概念速记
| 概念 | 要点 |
|------|------|
| **DESeq()** | 执行完整的差异分析流程(标准化→离散度估计→统计检验) |
| **results()** | 提取差异分析结果,返回DataFrame |
| **padj** | BH校正后的p值,用于差异基因筛选 |
| **log2FoldChange** | log2倍数变化,表示差异大小和方向 |
| **clusterProfiler** | 功能富集分析的R包,支持GO/KEGG/GSEA |
### ⚠️ 常见错误
| 错误 | 正确做法 |
|------|----------|
| 使用pvalue而非padj筛选 | 必须使用padj(BH校正后) |
| 仅看p值不看log2FC | 需同时满足padj<0.05和\|log2FC\|>1 |
| GO分析使用全部基因 | 背景应为所有检测到的基因 |
:::
---
## 实验准备
### 加载包和真实数据
```{r}
# 安装和加载所需包
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
packages <- c("DESeq2", "ggplot2", "pheatmap", "clusterProfiler",
"org.Hs.eg.db", "enrichplot")
for (pkg in packages) {
if (!require(pkg, character.only = TRUE, quietly = TRUE)) {
BiocManager::install(pkg)
library(pkg, character.only = TRUE)
}
}
# 中文字体
showtext::showtext_auto()
# 加载真实RNA-seq数据(PTC vs ATC甲状腺癌数据)
# 如果实验1已完成,可以直接使用dds对象
# 否则运行以下代码重新加载数据
# 数据下载链接(从课程网站下载):
# - geneCountMatrix.txt: ../data/geneCountMatrix.txt
# - samplesinfo.txt: ../data/samplesinfo.txt
# 代码中使用GitHub链接确保可执行
# 下载数据文件(如果不存在)
data_url_counts <- "http://biotree.top:38123/courses/rna-seq-analysis/data/geneCountMatrix.txt"
data_url_info <- "http://biotree.top:38123/courses/rna-seq-analysis/data/samplesinfo.txt"
#data_url_counts <- "https://github.com/WangLabCSU/courses/raw/main/rna-seq-analysis/data/geneCountMatrix.txt"
#data_url_info <- "https://github.com/WangLabCSU/courses/raw/main/rna-seq-analysis/data/samplesinfo.txt"
if (!file.exists("../data/geneCountMatrix.txt")) {
download.file(data_url_counts, "geneCountMatrix.txt", mode = "wb")
cat("已下载 geneCountMatrix.txt\n")
count_file <- "geneCountMatrix.txt"
} else {
count_file <- "../data/geneCountMatrix.txt"
}
if (!file.exists("../data/samplesinfo.txt")) {
download.file(data_url_info, "samplesinfo.txt", mode = "wb")
cat("已下载 samplesinfo.txt\n")
info_file <- "samplesinfo.txt"
} else {
info_file <- "../data/samplesinfo.txt"
}
# 1. 读取表达矩阵
counts_matrix <- read.table(count_file, header = TRUE, sep = "\t", check.names = FALSE)
counts <- counts_matrix[, -1] # 移除基因symbol列
rownames(counts) <- counts_matrix$gene_symbol
# 2. 读取样本信息
sample_info <- read.table(info_file, header = TRUE, sep = "\t")
conditions <- factor(sample_info$type, levels = c("PTC", "ATC"))
# 3. 创建DESeq2对象
colData <- data.frame(
condition = conditions,
batch = sample_info$batch,
row.names = sample_info$sample
)
dds <- DESeqDataSetFromMatrix(
countData = counts,
colData = colData,
design = ~ condition
)
# 4. 过滤低表达基因
keep <- rowSums(counts(dds) >= 10) >= 3
dds <- dds[keep, ]
cat("数据准备完成!\n")
cat("基因数:", nrow(dds), "\n")
cat("样本数:", ncol(dds), "\n")
cat("样本名称:", colnames(dds), "\n")
```
---
## 第一部分:差异表达分析
### 练习1.1:运行DESeq2差异分析
```{r}
# TODO: 完成DESeq2差异分析流程
# 1. 运行DESeq2(包含标准化、离散度估计、统计检验)
cat("运行DESeq2分析...\n")
dds <- DESeq(dds)
# 2. 提取结果
# contrast参数:c("分组列名", "处理组", "对照组")
# 注意:这里的"处理"是ATC vs PTC
res <- results(dds, contrast = c("condition", "ATC", "PTC"))
# 3. (可选) 收缩log2FC估计值,使低表达基因的fold change更稳健
# res_lfc <- lfcShrink(dds, coef = "condition_ATC_vs_PTC", type = "apeglm")
# 推荐使用收缩后的log2FC进行下游可视化
# 3. 查看结果摘要
cat("\nDESeq2结果摘要:\n")
summary(res)
# 4. 查看结果表的前几行
cat("\n结果表(前5行):\n")
print(head(res))
# 5. 统计各p值范围的基因数
cat("\np值分布:\n")
cat("padj < 0.001:", sum(res$padj < 0.001, na.rm = TRUE), "\n")
cat("padj < 0.01:", sum(res$padj < 0.01, na.rm = TRUE), "\n")
cat("padj < 0.05:", sum(res$padj < 0.05, na.rm = TRUE), "\n")
```
<details>
<summary>点击查看输出解读</summary>
**输出解读**:
- `baseMean`:平均表达量(size factor标准化后)
- `log2FoldChange`:log2倍数变化(ATC vs PTC)
- `lfcSE`:log2FC的标准误
- `stat`:Wald统计量
- `pvalue`:原始p值
- `padj`:BH校正后的p值
</details>
---
### 练习1.2:理解结果表并筛选差异基因
```{r}
# TODO: 整理和解读差异分析结果
# 1. 将结果转换为数据框并移除NA
res_df <- as.data.frame(res)
res_df$gene <- rownames(res_df)
res_df <- na.omit(res_df) # 移除padj为NA的行(通常是低表达基因被独立过滤)
# 2. 添加差异状态分类
res_df$diff_status <- ifelse(
res_df$padj < 0.05 & res_df$log2FoldChange > 1, "Up",
ifelse(res_df$padj < 0.05 & res_df$log2FoldChange < -1, "Down", "Not Sig")
)
# 3. 统计各类基因数量
status_table <- table(res_df$diff_status)
cat("差异基因统计:\n")
print(status_table)
# 4. 找出Top 10上调基因(按padj排序)
top_up <- res_df[res_df$diff_status == "Up", ]
top_up <- top_up[order(top_up$padj), ]
cat("\nTop 10 上调基因 (ATC中高表达):\n")
print(head(top_up[, c("gene", "baseMean", "log2FoldChange", "padj")], 10))
# 5. 找出Top 10下调基因
top_down <- res_df[res_df$diff_status == "Down", ]
top_down <- top_down[order(top_down$padj), ]
cat("\nTop 10 下调基因 (ATC中低表达):\n")
print(head(top_down[, c("gene", "baseMean", "log2FoldChange", "padj")], 10))
```
<details>
<summary>点击查看核心代码</summary>
```r
res_df <- as.data.frame(res)
res_df$gene <- rownames(res_df)
res_df <- na.omit(res_df)
res_df$diff_status <- ifelse(
res_df$padj < 0.05 & res_df$log2FoldChange > 1, "Up",
ifelse(res_df$padj < 0.05 & res_df$log2FoldChange < -1, "Down", "Not Sig")
)
table(res_df$diff_status)
```
</details>
::: {.callout-tip}
### 筛选阈值说明
- **padj < 0.05**:控制假发现率在5%以下
- **|log2FC| > 1**:倍数变化 > 2倍
- 可根据研究需要调整:严格(|log2FC|>2, padj<0.01)或宽松(|log2FC|>0.5, padj<0.1)
:::
---
## 第二部分:结果可视化
### 练习2.1:MA图
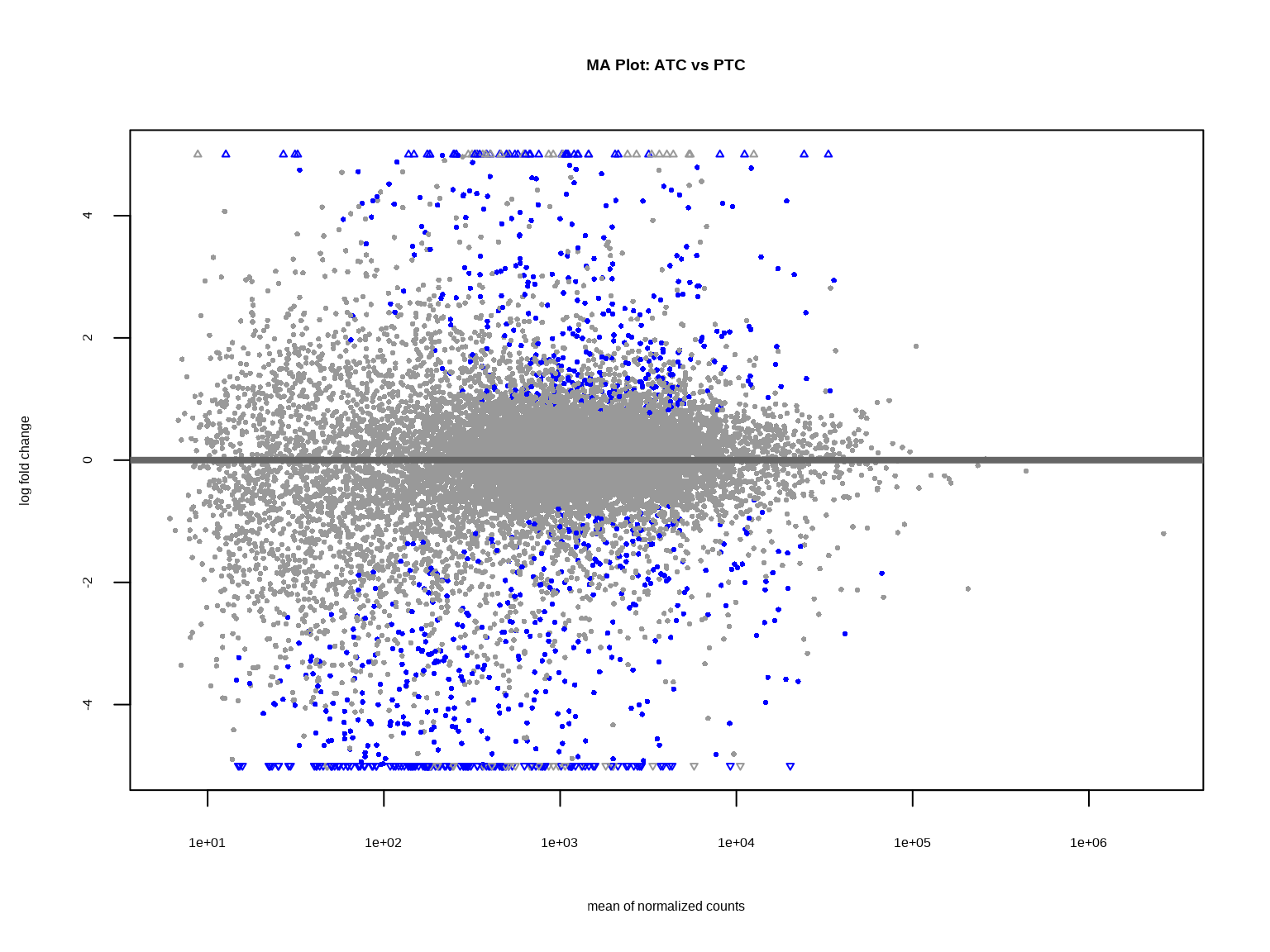
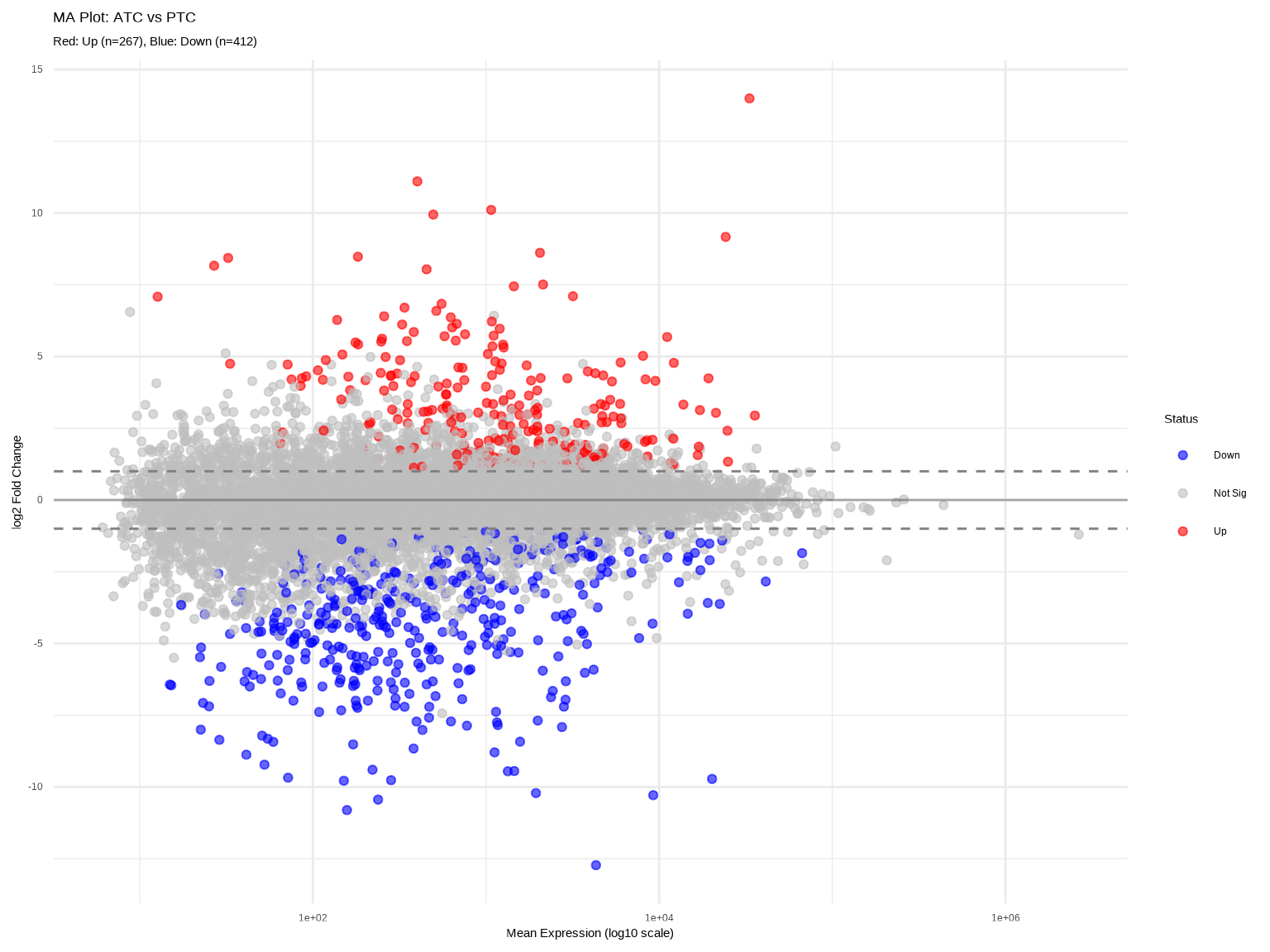
MA图展示表达量(mean)与差异倍数(log2FC)的关系。
```{r}
# TODO: 创建MA图
# 方法1:使用DESeq2内置函数(快速)
plotMA(res, ylim = c(-5, 5), main = "MA Plot: ATC vs PTC")
# 方法2:使用ggplot2自定义(更美观)
ma_plot <- ggplot(res_df, aes(x = baseMean, y = log2FoldChange, color = diff_status)) +
geom_point(alpha = 0.6, size = 1.5) +
scale_x_log10() +
scale_color_manual(values = c("Down" = "blue", "Not Sig" = "grey", "Up" = "red")) +
geom_hline(yintercept = c(-1, 1), linetype = "dashed", color = "grey50") +
geom_hline(yintercept = 0, linetype = "solid", color = "black", alpha = 0.3) +
theme_minimal() +
labs(title = "MA Plot: ATC vs PTC",
subtitle = paste0("Red: Up (n=", sum(res_df$diff_status=="Up"),
"), Blue: Down (n=", sum(res_df$diff_status=="Down"), ")"),
x = "Mean Expression (log10 scale)",
y = "log2 Fold Change",
color = "Status")
print(ma_plot)
```
<details>
<summary>点击查看MA图解读</summary>
**MA图解读**:
- **X轴**:平均表达量(log10)
- **Y轴**:log2 Fold Change
- **红点**:上调基因(ATC中高表达)
- **蓝点**:下调基因(ATC中低表达)
- **灰点**:不显著
**关键观察**:
- 低表达基因(左侧)的log2FC波动更大
- 高表达基因(右侧)趋于稳定
- 这是RNA-seq数据的典型特征
</details>
---
### 练习2.2:火山图
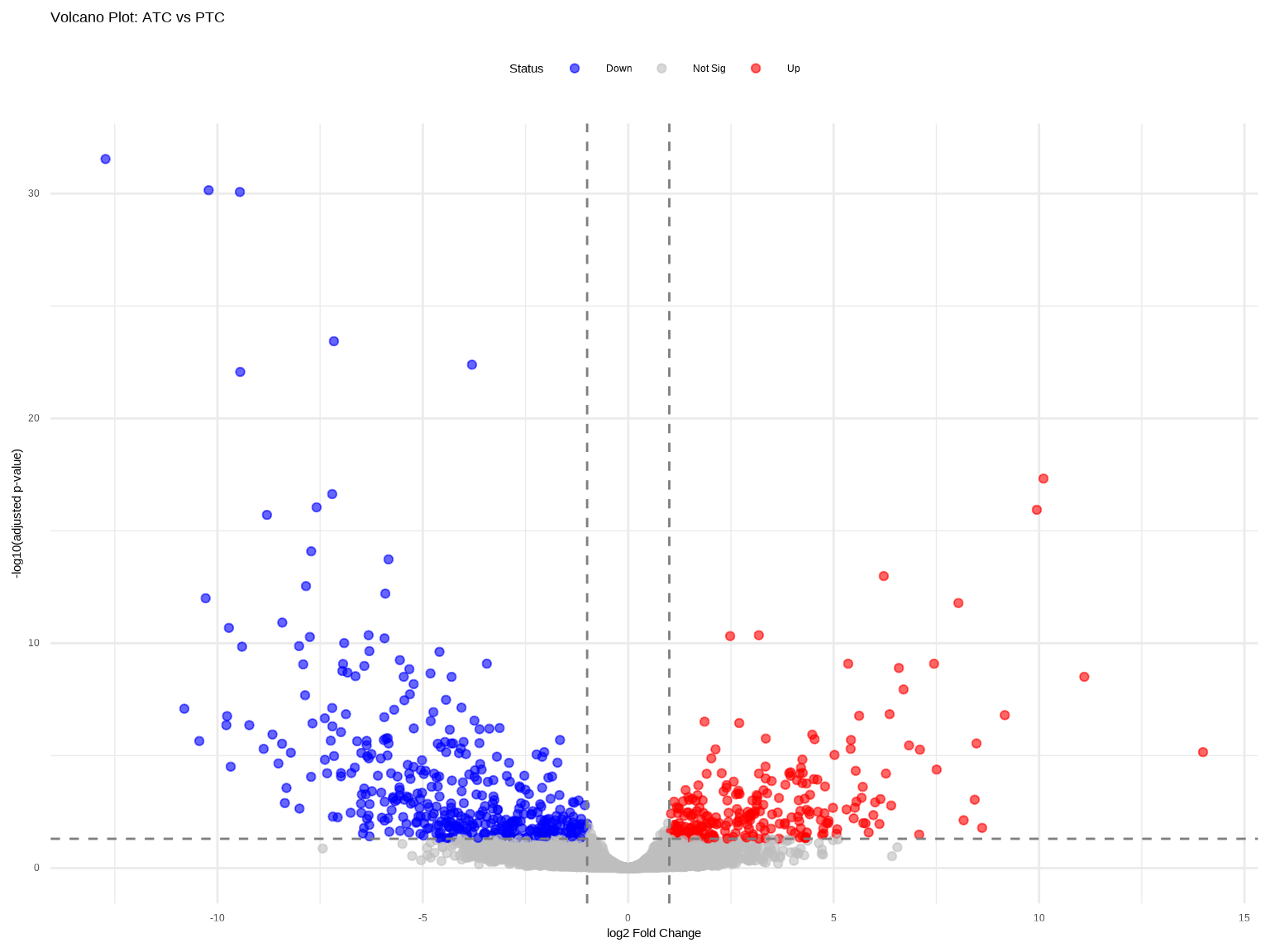
火山图同时展示差异倍数和统计显著性。
```{r}
# TODO: 创建火山图
# 1. 准备数据
res_plot <- res_df
res_plot$negLog10padj <- -log10(res_plot$padj)
# 2. 绘制火山图
volcano_plot <- ggplot(res_plot, aes(x = log2FoldChange, y = negLog10padj,
color = diff_status)) +
geom_point(alpha = 0.6, size = 1.5) +
scale_color_manual(values = c("Down" = "blue", "Not Sig" = "grey", "Up" = "red")) +
geom_vline(xintercept = c(-1, 1), linetype = "dashed", color = "grey50") +
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "grey50") +
theme_minimal() +
labs(title = "Volcano Plot: ATC vs PTC",
x = "log2 Fold Change",
y = "-log10(adjusted p-value)",
color = "Status") +
theme(legend.position = "top")
print(volcano_plot)
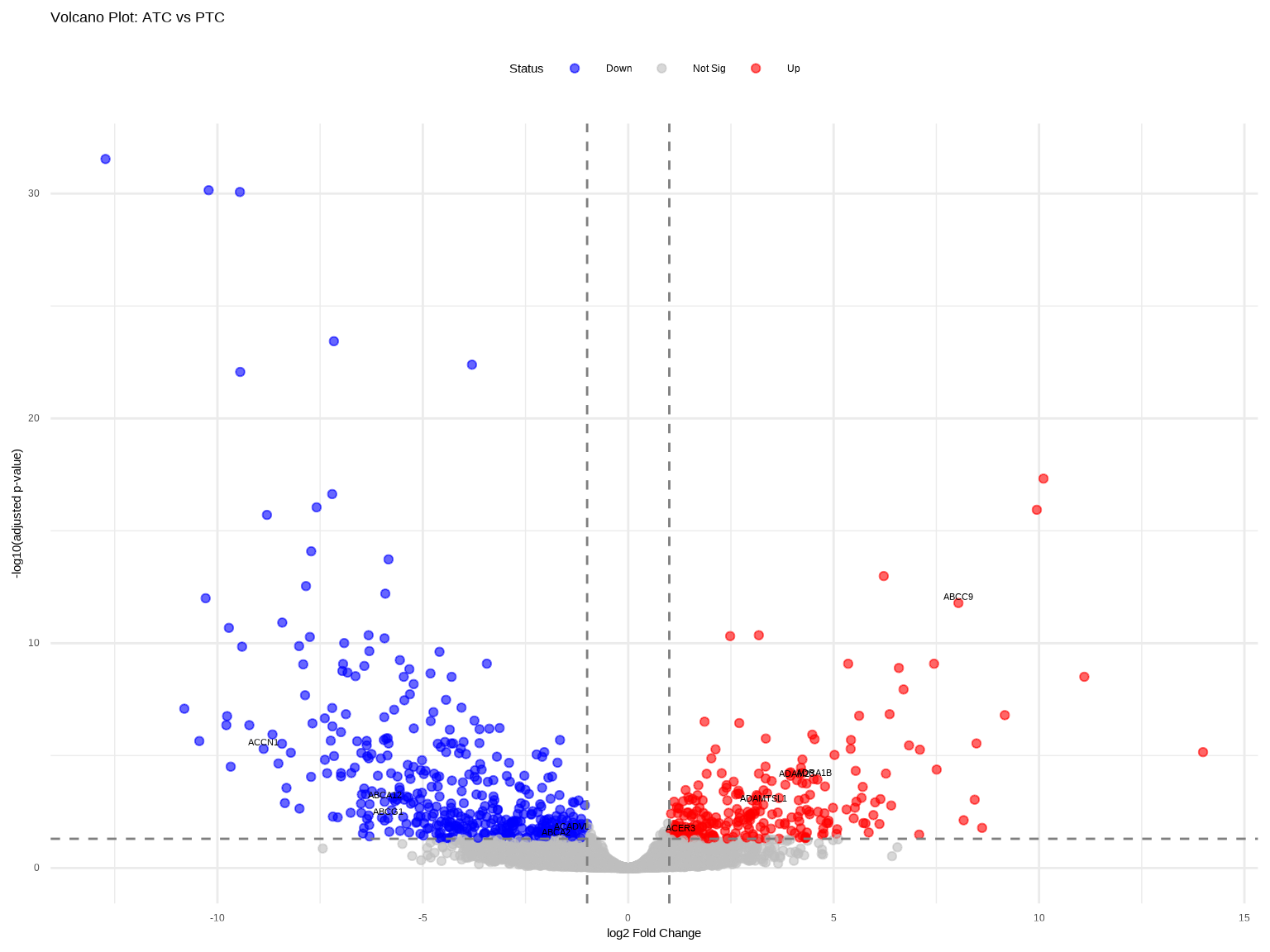
# 3. 标注Top基因(可选)
# 获取最显著的上下调基因各5个
top_genes <- rbind(
head(res_plot[res_plot$diff_status == "Up", ], 5),
head(res_plot[res_plot$diff_status == "Down", ], 5)
)
volcano_labeled <- volcano_plot +
geom_text(data = top_genes,
aes(label = gene),
size = 3, vjust = -0.5, hjust = 0.5, color = "black")
print(volcano_labeled)
```
<details>
<summary>点击查看核心代码</summary>
```r
res_plot <- res_df
res_plot$negLog10padj <- -log10(res_plot$padj)
volcano_plot <- ggplot(res_plot, aes(x = log2FoldChange, y = negLog10padj,
color = diff_status)) +
geom_point(alpha = 0.6, size = 1.5) +
scale_color_manual(values = c("Down" = "blue", "Not Sig" = "grey", "Up" = "red")) +
geom_vline(xintercept = c(-1, 1), linetype = "dashed", color = "grey50") +
geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "grey50") +
theme_minimal() +
labs(title = "Volcano Plot: ATC vs PTC",
x = "log2 Fold Change",
y = "-log10(adjusted p-value)")
print(volcano_plot)
```
</details>
::: {.callout-note}
### 火山图解读
- **左上/右上区域**:既显著又差异大的基因
- **顶部水平线**:padj = 0.05阈值
- **垂直虚线**:|log2FC| = 1阈值
- **注意**:|log2FC|很大但p值不显著的点(左右两端灰色点)通常是低表达基因
:::
---
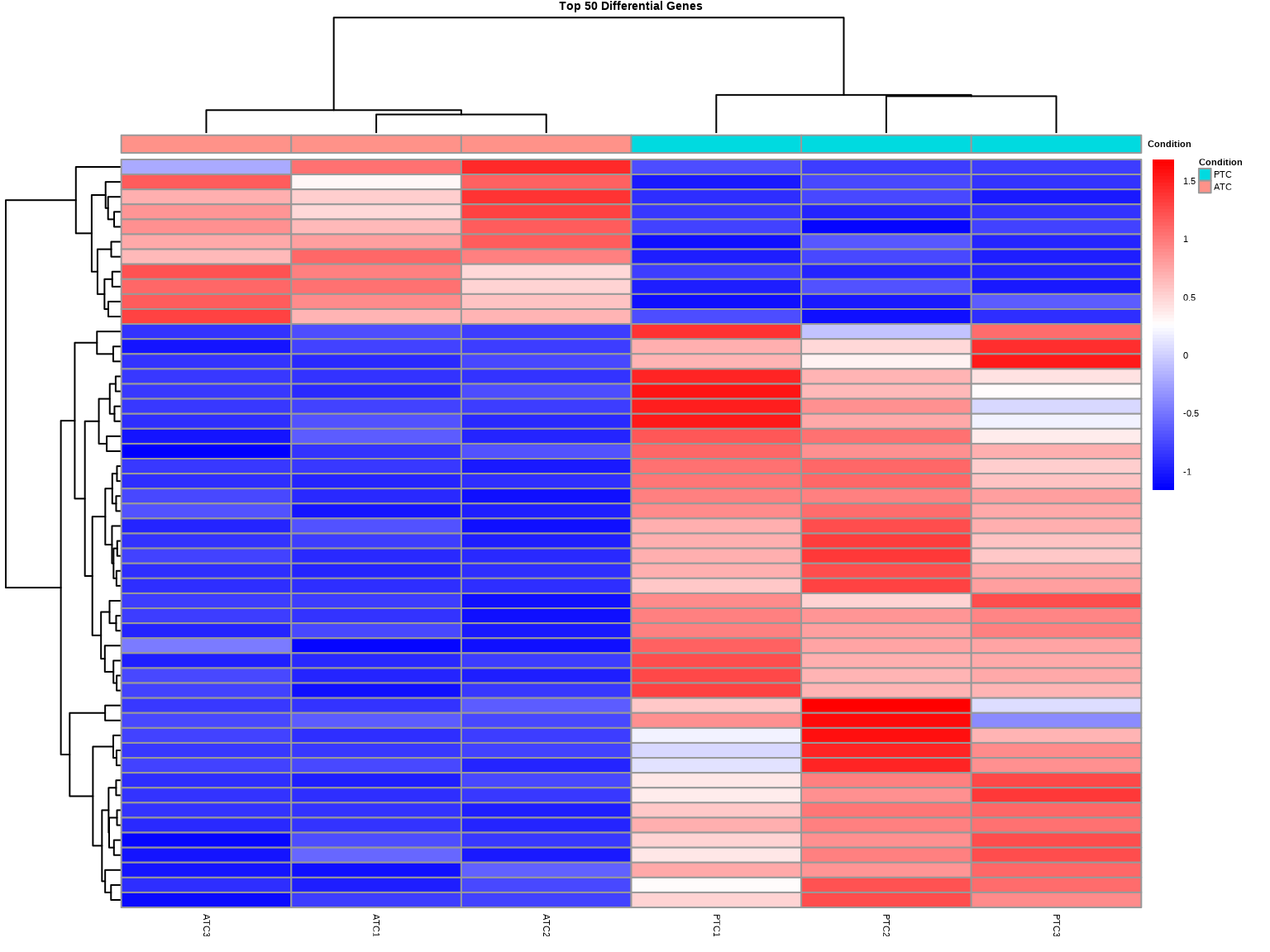
### 练习2.3:差异基因热图
```{r}
# TODO: 创建差异基因表达热图
# 1. 选择Top差异基因进行可视化(padj最小的前50个)
top_genes_heatmap <- head(res_df[order(res_df$padj), "gene"], 50)
# 2. 获取VST转换后的数据
dds_vst <- vst(dds, blind = FALSE)
vst_matrix <- assay(dds_vst)
# 提取Top基因的表达量
heatmap_data <- vst_matrix[top_genes_heatmap, ]
# 3. 按行(基因)标准化,突出表达模式
heatmap_scaled <- t(scale(t(heatmap_data)))
# 4. 准备注释数据
annotation_col <- data.frame(
Condition = colData(dds)$condition,
row.names = colnames(dds)
)
# 5. 绘制热图
pheatmap(heatmap_scaled,
annotation_col = annotation_col,
cluster_rows = TRUE,
cluster_cols = TRUE,
show_rownames = FALSE,
main = "Top 50 Differential Genes",
color = colorRampPalette(c("blue", "white", "red"))(100),
fontsize = 8)
```
<details>
<summary>点击查看核心代码</summary>
```r
top_genes_heatmap <- head(res_df[order(res_df$padj), "gene"], 50)
dds_vst <- vst(dds, blind = FALSE)
vst_matrix <- assay(dds_vst)
heatmap_data <- vst_matrix[top_genes_heatmap, ]
heatmap_scaled <- t(scale(t(heatmap_data)))
annotation_col <- data.frame(
Condition = colData(dds)$condition,
row.names = colnames(dds)
)
pheatmap(heatmap_scaled,
annotation_col = annotation_col,
cluster_rows = TRUE,
cluster_cols = TRUE,
show_rownames = FALSE,
main = "Top 50 Differential Genes",
color = colorRampPalette(c("blue", "white", "red"))(100))
```
</details>
::: {.callout-tip}
### 热图解读
- **红色**:高表达
- **蓝色**:低表达
- **列聚类**:样本是否按条件分组
- **行标准化**:消除基因间基线差异,突出相对变化
:::
---
## 第三部分:功能富集分析
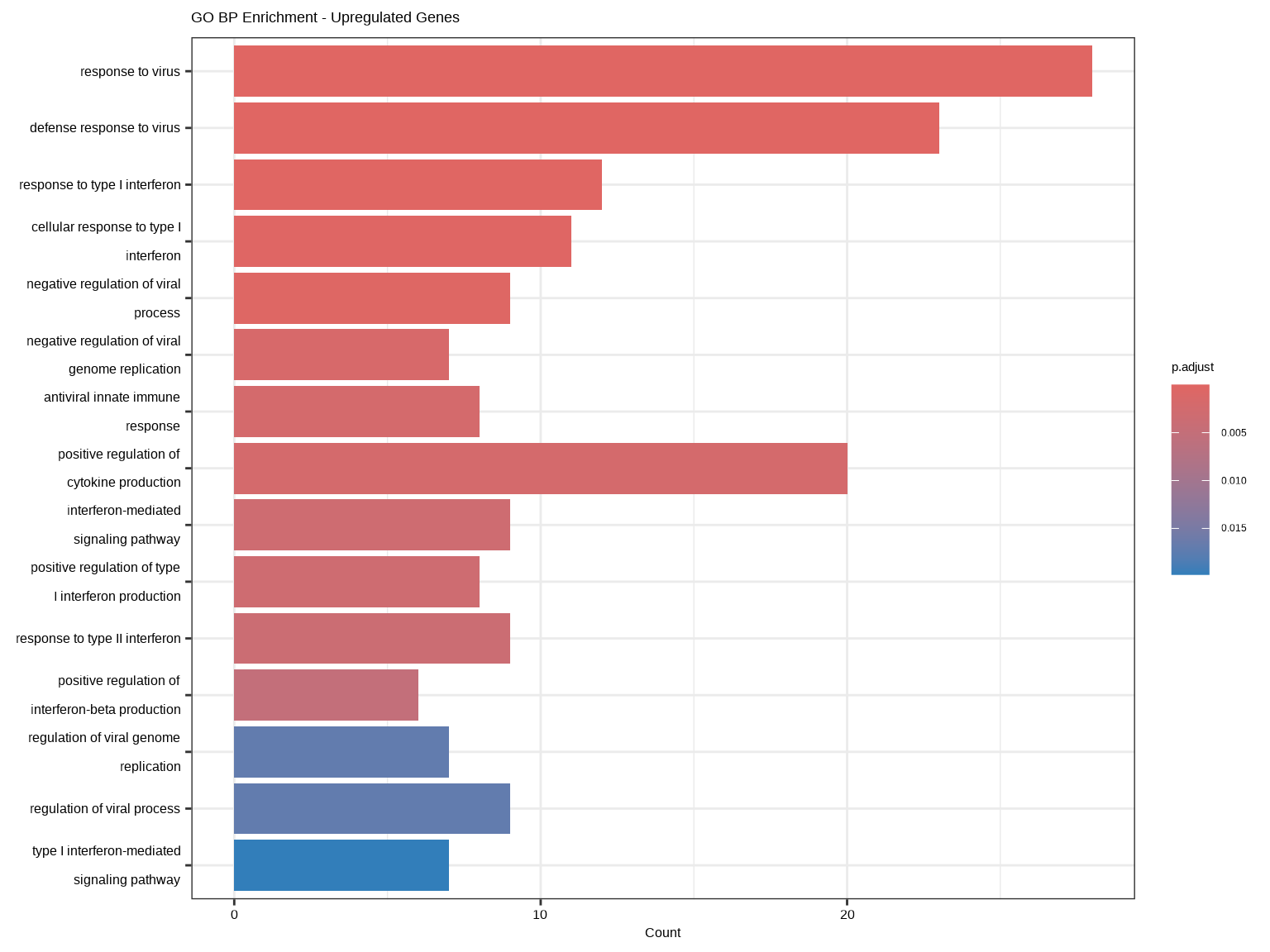
### 练习3.1:GO富集分析
```{r}
# TODO: 进行GO功能富集分析
# 1. 准备差异基因列表
gene_list_up <- res_df$gene[res_df$diff_status == "Up"]
gene_list_down <- res_df$gene[res_df$diff_status == "Down"]
cat("上调基因数:", length(gene_list_up), "\n")
cat("下调基因数:", length(gene_list_down), "\n")
# 2. 查看支持的基因ID类型
cat("\norg.Hs.eg.db支持的ID类型(部分):\n")
print(head(keytypes(org.Hs.eg.db), 10))
# 3. GO富集分析(Biological Process)
# 使用SYMBOL直接分析(clusterProfiler支持)
ego_up <- enrichGO(
gene = gene_list_up,
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL", # 输入基因ID类型
ont = "BP", # BP: Biological Process
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
qvalueCutoff = 0.2
)
# 4. 查看结果
cat("\nGO富集结果(前10条):\n")
print(head(ego_up, 10))
# 5. 可视化
# 条形图
barplot(ego_up, showCategory = 15, title = "GO BP Enrichment - Upregulated Genes")
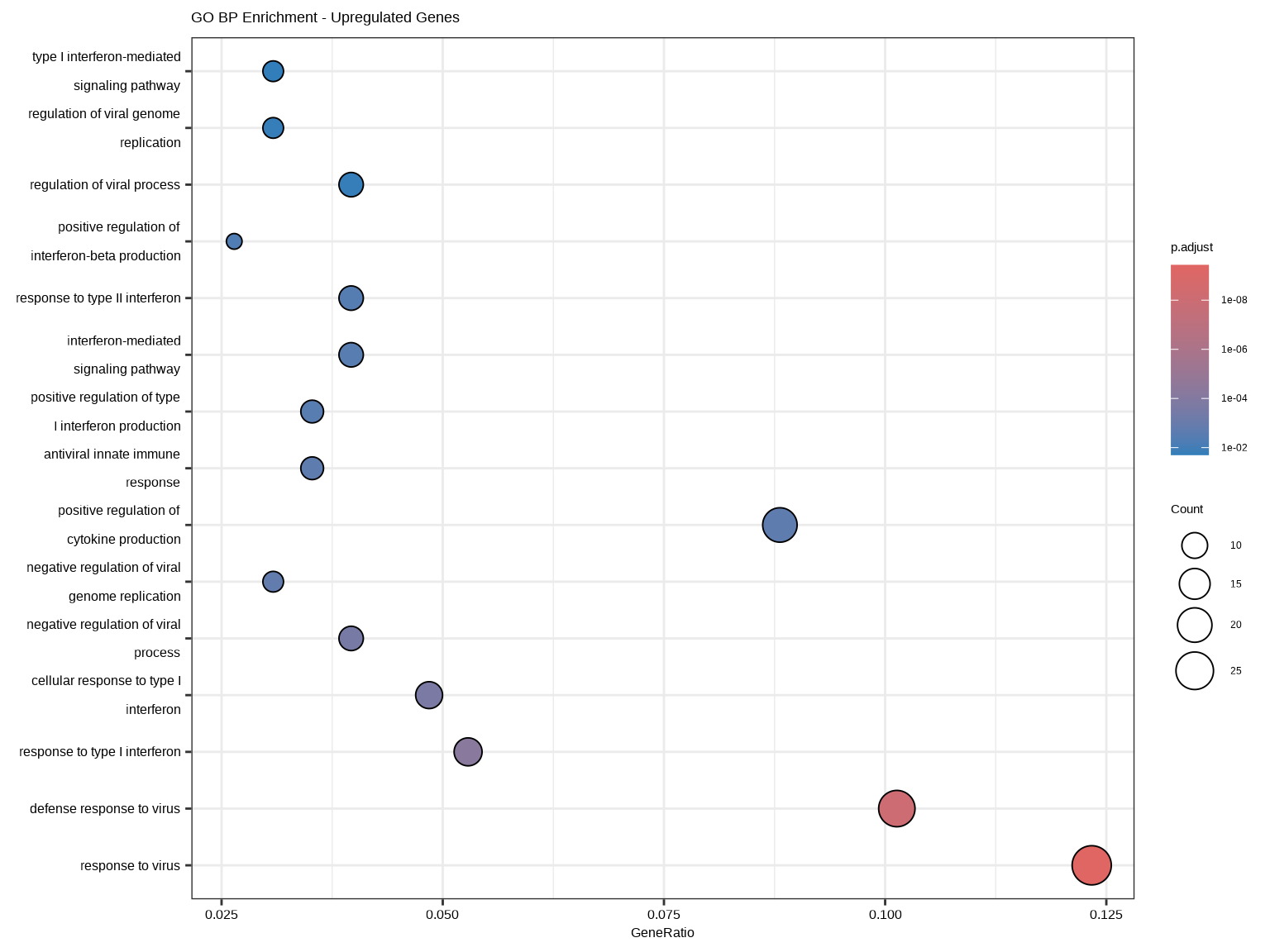
# 点图(更推荐使用)
dotplot(ego_up, showCategory = 15, orderBy = "p.adjust",
title = "GO BP Enrichment - Upregulated Genes")
# 6. 保存结果
write.csv(as.data.frame(ego_up), "GO_BP_upregulated.csv", row.names = FALSE)
cat("\n结果已保存到: GO_BP_upregulated.csv\n")
```
<details>
<summary>点击查看结果解读</summary>
**富集结果解读**:
- **ID**:GO条目ID
- **Description**:功能描述
- **GeneRatio**:差异基因中属于该通路的基因比例
- **BgRatio**:背景基因中属于该通路的基因比例
- **pvalue/p.adjust**:富集显著性
- **qvalue**:FDR控制
- **Count**:属于该通路的差异基因数
</details>
::: {.callout-warning}
### 富集分析注意事项
1. **背景基因集**:应为所有检测到的基因,而非全基因组
2. **基因ID转换**:clusterProfiler需要特定ID格式(ENTREZID/SYMBOL等)
3. **阈值选择**:p.adjust < 0.05为常用阈值
4. **生物学解释**:富集≠因果,需结合文献解读
5. **GO冗余**:GO terms常有高度重叠,可使用 `simplify(ego)` 减少冗余
:::
---
### 练习3.2:KEGG通路富集分析
```{r}
# TODO: 进行KEGG通路富集分析
# 1. 基因ID转换(SYMBOL → ENTREZID)
gene_df <- bitr(gene_list_up,
fromType = "SYMBOL",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
cat("成功转换的基因数:", nrow(gene_df), "\n")
# 2. KEGG富集分析
kk <- enrichKEGG(
gene = gene_df$ENTREZID,
organism = "hsa", # hsa = human
pAdjustMethod = "BH",
pvalueCutoff = 0.05
)
# 3. 查看结果
cat("\nKEGG富集结果:\n")
print(head(kk, 10))
# 4. 可视化
dotplot(kk, showCategory = 15, title = "KEGG Pathway Enrichment")
```
<details>
<summary>点击查看核心代码</summary>
```r
gene_df <- bitr(gene_list_up, fromType = "SYMBOL",
toType = "ENTREZID", OrgDb = org.Hs.eg.db)
kk <- enrichKEGG(gene = gene_df$ENTREZID, organism = "hsa",
pAdjustMethod = "BH", pvalueCutoff = 0.05)
dotplot(kk, showCategory = 15)
```
</details>
::: {.callout-note}
### 常用物种代码
| 物种 | 代码 |
|------|------|
| 人类 | hsa |
| 小鼠 | mmu |
| 大鼠 | rno |
| 斑马鱼 | dre |
:::
---
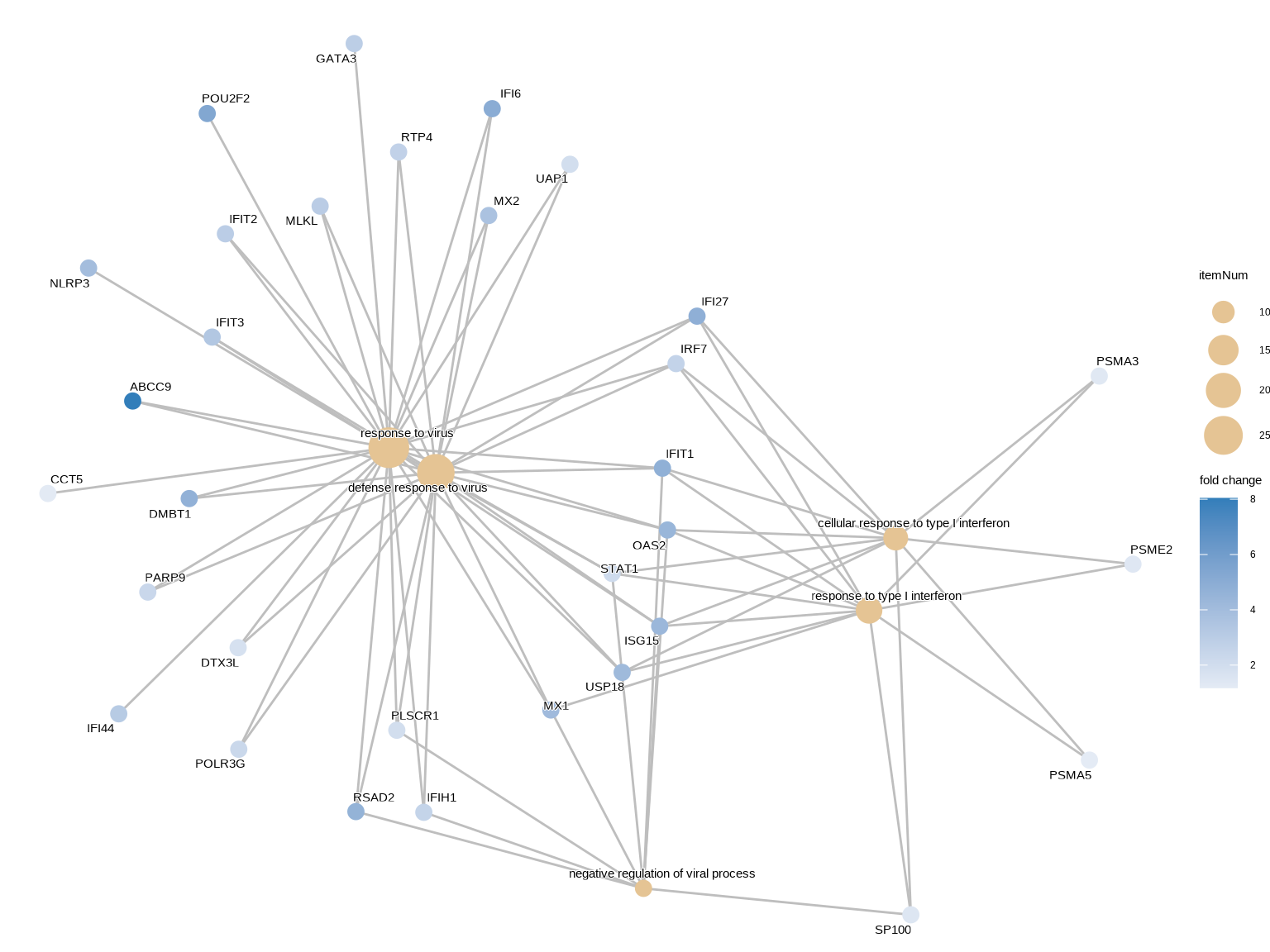
### 练习3.3:网络图可视化(cnetplot)
```{r}
# TODO: 绘制富集基因与通路的网络图
# 1. 准备基因表达变化数据
gene_foldchange <- res_df$log2FoldChange
names(gene_foldchange) <- res_df$gene
# 2. 绘制cnetplot
# 显示前5个显著富集的条目及其关联基因
cnetplot(ego_up,
foldChange = gene_foldchange,
showCategory = 5 # 显示前5个条目
)
```
<details>
<summary>点击查看cnetplot解读</summary>
**cnetplot解读**:
- **大节点**:富集条目(GO term)
- **小节点**:基因
- **用途**:查看哪些基因参与多个通路,以及基因与通路的关联关系
</details>
---
### 练习3.4:GSEA分析(可选,进阶)
```{r}
# TODO: 进行GSEA分析
# 1. 准备排序基因列表(所有基因,按log2FC排序)
geneList <- res_df$log2FoldChange
names(geneList) <- res_df$gene
geneList <- sort(geneList, decreasing = TRUE)
# 2. 查看排序后的基因列表
cat("排序后的基因列表(Top 10上调和下调):\n")
cat("Top 10 上调基因:\n")
print(head(geneList, 10))
cat("\nTop 10 下调基因:\n")
print(tail(geneList, 10))
# 3. GSEA分析(使用SYMBOL)
gsea <- gseGO(
geneList = geneList,
OrgDb = org.Hs.eg.db,
keyType = "SYMBOL",
ont = "BP",
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
minGSSize = 10, # 基因集最小基因数
maxGSSize = 500 # 基因集最大基因数
)
# 4. 查看GSEA结果
cat("\nGSEA结果(前5条):\n")
print(head(gsea, 5))
# 5. GSEA可视化
# 绘制最显著富集的条目
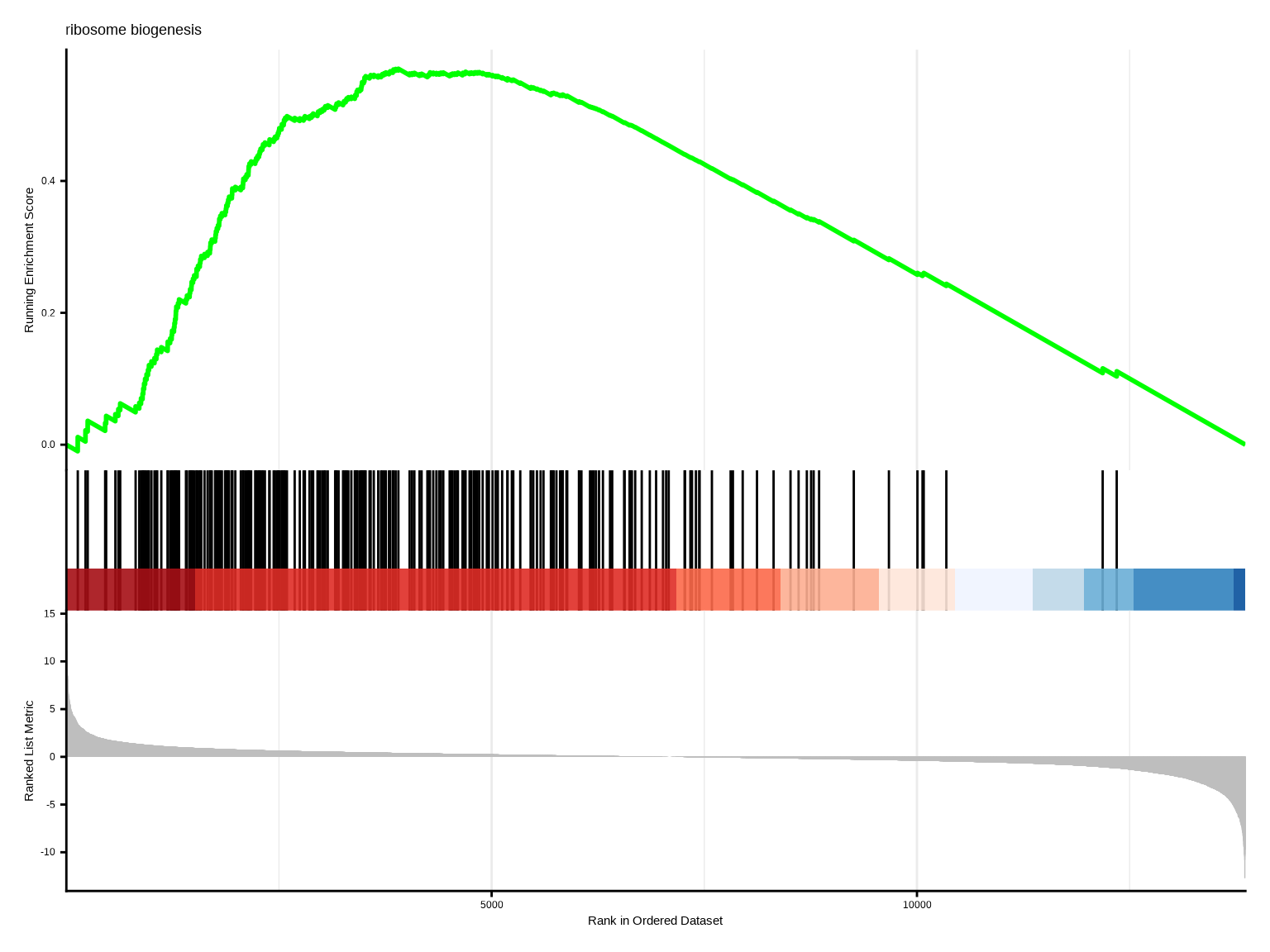
gseaplot2(gsea, geneSetID = 1, title = gsea$Description[1])
# 同时绘制前3个
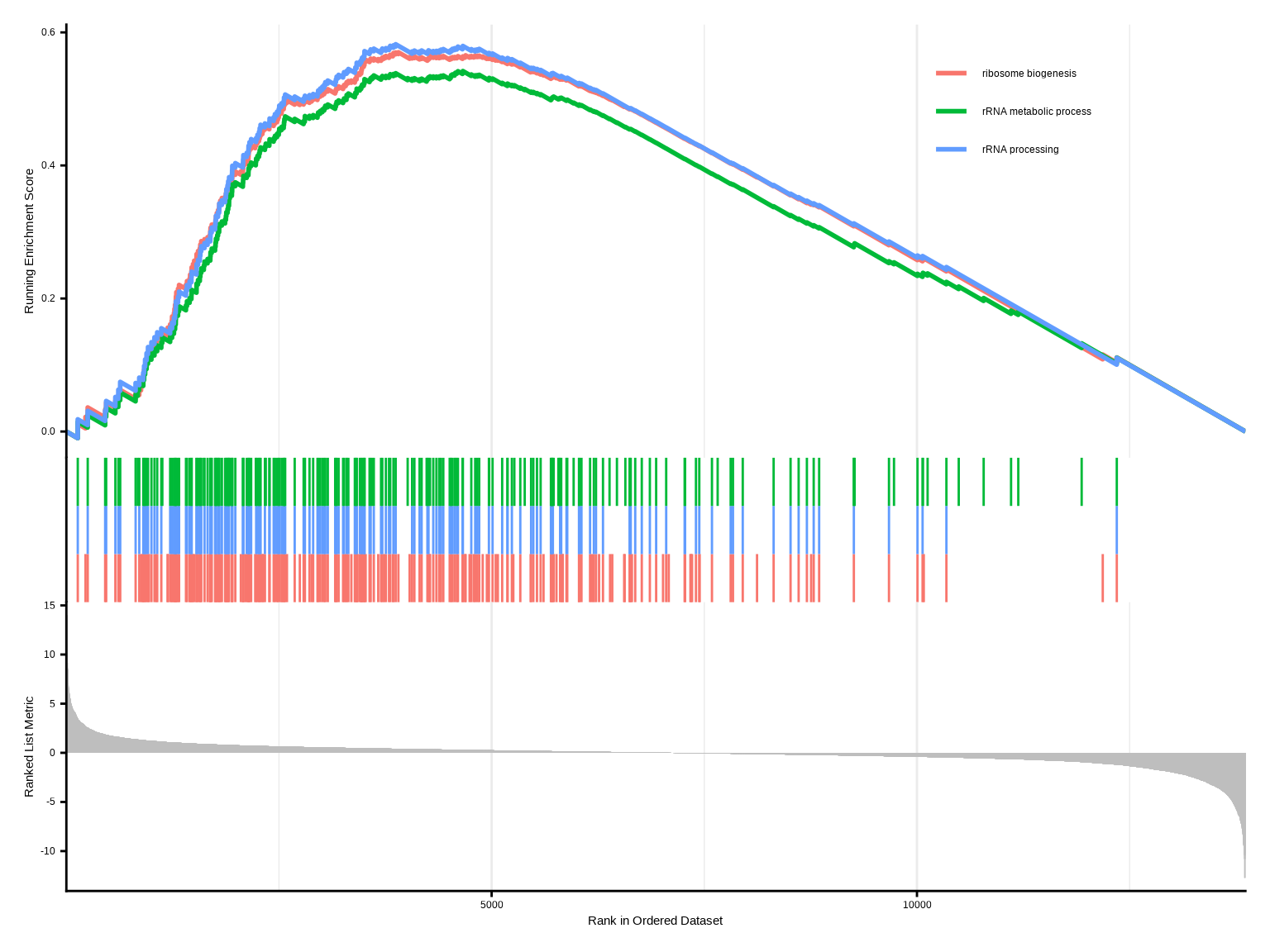
gseaplot2(gsea, geneSetID = 1:3)
```
<details>
<summary>点击查看GSEA解读</summary>
**GSEA结果解读**:
- **NES**:标准化富集分数
- NES > 0:通路在排序列表顶部富集(上调)
- NES < 0:通路在排序列表底部富集(下调)
- **p.adjust**:校正后的显著性
- **leading_edge**:核心富集基因
**GSEA mountain plot解读**:
- 绿线:富集分数曲线,峰值位置表示最大富集
- 黑色竖线:基因集中基因在排序列表中的位置
- 热图:基因表达值分布
</details>
---
## 第四部分:综合分析报告
### 练习4.1:生成差异分析报告
```r
# TODO: 生成差异分析汇总报告
cat("==============================================\n")
cat(" RNA-seq 差异分析报告\n")
cat(" PTC vs ATC\n")
cat("==============================================\n\n")
cat("1. 样本信息\n")
cat(" - 对照组(PTC)样本数:", sum(colData$condition == "PTC"), "\n")
cat(" - 处理组(ATC)样本数:", sum(colData$condition == "ATC"), "\n")
cat(" - 总基因数:", nrow(res_df), "\n\n")
cat("2. 差异基因统计\n")
cat(" - 总差异基因数:", sum(res_df$diff_status != "Not Sig"), "\n")
cat(" - 上调基因数(ATC高):", sum(res_df$diff_status == "Up"), "\n")
cat(" - 下调基因数(ATC低):", sum(res_df$diff_status == "Down"), "\n\n")
cat("3. Top 5 上调基因\n")
top5_up <- head(res_df[res_df$diff_status == "Up", ], 5)
for (i in 1:5) {
cat(sprintf(" %s: log2FC = %.2f, padj = %.2e\n",
top5_up$gene[i], top5_up$log2FoldChange[i], top5_up$padj[i]))
}
cat("\n4. Top 5 下调基因\n")
top5_down <- head(res_df[res_df$diff_status == "Down", ], 5)
for (i in 1:5) {
cat(sprintf(" %s: log2FC = %.2f, padj = %.2e\n",
top5_down$gene[i], top5_down$log2FoldChange[i], top5_down$padj[i]))
}
cat("\n5. 功能富集分析结果\n")
if (exists("ego_up") && nrow(as.data.frame(ego_up)) > 0) {
cat(" - GO BP富集条目数:", nrow(as.data.frame(ego_up)), "\n")
cat(" - Top富集通路:", as.data.frame(ego_up)$Description[1], "\n")
} else {
cat(" - GO BP富集条目数: 0(可能需要调整阈值)\n")
}
if (exists("kk") && !is.null(kk) && nrow(as.data.frame(kk)) > 0) {
cat(" - KEGG富集条目数:", nrow(as.data.frame(kk)), "\n")
} else {
cat(" - KEGG富集条目数: 0或分析未完成\n")
}
cat("\n6. 生物学解释\n")
cat(" - 上调基因可能参与ATC的恶性表型\n")
cat(" - 下调基因可能在ATC中受到抑制\n")
cat(" - 富集到的通路提示了ATC与PTC的生物学差异\n")
cat(" - 需结合文献进一步验证关键通路\n")
cat("\n==============================================\n")
```
---
## 思考题
1. **为什么火山图中会出现|log2FC|很大但p值不显著的点?**
<details>
<summary>点击查看答案</summary>
这些基因通常表达量很低,counts的随机波动大,统计检验效能不足
</details>
2. **MA图中为什么高表达基因的log2FC趋于0?**
<details>
<summary>点击查看答案</summary>
高表达基因通常受严格调控,表达量相对稳定;低表达基因更容易出现大倍数变化
</details>
3. **如何选择差异基因筛选阈值?**
<details>
<summary>点击查看答案</summary>
标准:|log2FC| > 1, padj < 0.05。可根据研究需要调整,严格阈值用于下游验证,宽松阈值用于探索
</details>
4. **热图中为什么要对数据进行行标准化?**
<details>
<summary>点击查看答案</summary>
消除基因间表达量基线差异,突出展示基因在不同样本间的表达模式(相对变化)
</details>
5. **富集分析中为什么要进行多重检验校正?**
<details>
<summary>点击查看答案</summary>
检测大量GO/通路时,假阳性会累积,需用BH等方法控制FDR
</details>
---
## 关键代码速查表
| 操作 | 代码 |
|------|------|
| 运行DESeq2 | `dds <- DESeq(dds)` |
| 提取结果 | `res <- results(dds, contrast = c("condition", "A", "B"))` |
| 筛选差异基因 | `subset(res, padj < 0.05 & abs(log2FC) > 1)` |
| MA图 | `plotMA(res)` |
| 火山图 | `ggplot(res_df, aes(x=log2FC, y=-log10(padj), color=status)) + geom_point()` |
| 查看keytypes | `keytypes(org.Hs.eg.db)` |
| ID转换 | `bitr(genes, fromType="SYMBOL", toType="ENTREZID", OrgDb=org.Hs.eg.db)` |
| GO富集 | `enrichGO(gene, OrgDb=org.Hs.eg.db, ont="BP", keyType="SYMBOL")` |
| KEGG富集 | `enrichKEGG(gene, organism="hsa")` |
| 点图 | `dotplot(ego, showCategory=15, orderBy="p.adjust")` |
| 网络图 | `cnetplot(ego, foldChange=geneList, showCategory=5)` |
| GSEA | `gseGO(geneList, OrgDb=org.Hs.eg.db, ont="BP")` |
| GSEA图 | `gseaplot2(gsea, geneSetID=1)` |
---
## 延伸阅读
- [DESeq2 Vignette](https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html)
- [clusterProfiler Book](https://yulab-smu.top/biomedical-knowledge-mining-book/)
- [RNA-seq Visualization Guide](https://hbctraining.github.io/DGE_workshop/lessons/06_DGE_visualizing_results.html)
---
**实验结束,祝学习愉快!** 🎉