---
title: "实验1:RNA-seq数据质控与预处理"
subtitle: "从表达矩阵到质控报告"
execute:
freeze: false
eval: true
echo: true
output: true
warning: false
message: false
---
## 实验目标
完成本实验后,你将能够:
1. 使用DESeq2创建分析对象并进行数据预处理
2. 理解并计算Size Factors进行标准化
3. 使用VST进行数据转换
4. 通过PCA和相关性热图评估样本质量
5. 识别潜在的批次效应和异常样本
- **实验时长**:45分钟
- **实验数据**:基于真实甲状腺癌RNA-seq数据(PTC vs ATC)的简化数据集
- **数据下载**:<a href="../data/geneCountMatrix.txt" download>geneCountMatrix.txt</a> | <a href="../data/samplesinfo.txt" download>samplesinfo.txt</a>
---
## 关键知识点
::: {.callout-important}
### 核心概念速记
| 概念 | 要点 |
|------|------|
| **DESeqDataSet** | DESeq2的核心数据结构,包含counts矩阵、样本信息和设计公式 |
| **Size Factor** | 校正测序深度的标准化因子,中位数比值法计算 |
| **VST** | Variance Stabilizing Transformation,用于PCA和可视化 |
| **PCA** | 主成分分析,用于识别样本间主要变异来源 |
:::
---
## 实验准备
### 环境要求
- [R](https://cran.r-project.org/) >= 4.3.0
- 必需R包:DESeq2, ggplot2, pheatmap, RColorBrewer
### 安装和加载包
```{r}
# 安装Bioconductor包(如果尚未安装)
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
if (!require("DESeq2", quietly = TRUE))
BiocManager::install("DESeq2")
# 安装CRAN包
install_if_missing <- function(pkg) {
if (!require(pkg, character.only = TRUE, quietly = TRUE)) {
install.packages(pkg)
library(pkg, character.only = TRUE)
}
}
install_if_missing("ggplot2")
install_if_missing("pheatmap")
install_if_missing("RColorBrewer")
install_if_missing("reshape2")
# 加载包
library(DESeq2)
library(ggplot2)
library(pheatmap)
library(RColorBrewer)
# 中文字体
showtext::showtext_auto()
```
---
## 第一部分:数据准备
### 练习1.1:读取真实RNA-seq表达矩阵
我们将使用真实的甲状腺癌RNA-seq数据(PTC vs ATC)进行实验。数据已经过预处理,包含了基因表达计数矩阵和样本信息。
::: {.callout-note}
### 📦 数据集说明
| 文件 | 内容 | 样本 |
|------|------|:----:|
| `geneCountMatrix.txt` | 基因表达原始计数矩阵 | 6个样本 |
| `samplesinfo.txt` | 样本元数据(分组、批次) | 6行 |
**分组设计**:
- **PTC**(乳头状甲状腺癌):PTC1, PTC2, PTC3
- **ATC**(未分化甲状腺癌):ATC1, ATC2, ATC3
- **批次**:C1(ATC1, ATC3, PTC1),C2(ATC2, PTC2, PTC3)
**数据下载链接**:
- <a href="../data/geneCountMatrix.txt" download>geneCountMatrix.txt</a>
- <a href="../data/samplesinfo.txt" download>samplesinfo.txt</a>
:::
```{r}
# TODO: 下载数据文件(从课程网站下载到工作目录)
# 如果在本地运行,可以直接使用课程网站下载的文件
# 或从以下链接下载:
# - geneCountMatrix.txt: https://github.com/WangLabCSU/courses/raw/main/rna-seq-analysis/data/geneCountMatrix.txt
# - samplesinfo.txt: https://github.com/WangLabCSU/courses/raw/main/rna-seq-analysis/data/samplesinfo.txt
# 下载数据文件(如果不存在)
data_url_counts <- "http://biotree.top:38123/courses/rna-seq-analysis/data/geneCountMatrix.txt"
data_url_info <- "http://biotree.top:38123/courses/rna-seq-analysis/data/samplesinfo.txt"
#data_url_counts <- "https://github.com/WangLabCSU/courses/raw/main/rna-seq-analysis/data/geneCountMatrix.txt"
#data_url_info <- "https://github.com/WangLabCSU/courses/raw/main/rna-seq-analysis/data/samplesinfo.txt"
if (!file.exists("../data/geneCountMatrix.txt")) {
download.file(data_url_counts, "geneCountMatrix.txt", mode = "wb")
cat("已下载 geneCountMatrix.txt\n")
count_file <- "geneCountMatrix.txt"
} else {
count_file <- "../data/geneCountMatrix.txt"
}
if (!file.exists("../data/samplesinfo.txt")) {
download.file(data_url_info, "samplesinfo.txt", mode = "wb")
cat("已下载 samplesinfo.txt\n")
info_file <- "samplesinfo.txt"
} else {
info_file <- "../data/samplesinfo.txt"
}
# 3. 读取表达矩阵(基因计数矩阵)
counts_matrix <- read.table(count_file, header = TRUE, sep = "\t", check.names = FALSE)
# 查看数据结构
cat("原始数据维度:", dim(counts_matrix), "\n")
cat("\n前5行数据:\n")
print(head(counts_matrix, 5))
# 4. 设置行名为基因symbol,并移除基因symbol列
counts <- counts_matrix[, -1] # 移除第一列(基因symbol列)
rownames(counts) <- counts_matrix$gene_symbol
cat("\n表达矩阵维度:", dim(counts), "\n")
cat("样本名称:\n")
print(colnames(counts))
# 5. 读取样本信息
sample_info <- read.table(info_file, header = TRUE, sep = "\t")
cat("\n样本信息:\n")
print(sample_info)
# 6. 提取条件信息(PTC vs ATC)
# 注意:设置因子水平,PTC作为对照(参考水平)
conditions <- factor(sample_info$type, levels = c("PTC", "ATC"))
cat("\n条件因子:\n")
print(conditions)
# 7. 查看数据基本信息
cat("\n各样本总reads数(测序深度):\n")
print(colSums(counts))
cat("\n表达量分布统计:\n")
print(summary(as.vector(as.matrix(counts))))
cat("\n零值比例:", round(mean(counts == 0) * 100, 1), "%\n")
```
<details>
<summary>点击查看输出解读</summary>
**输出解读**:
- 矩阵维度:约20,000+个基因 × 6个样本(真实数据规模)
- 样本:ATC1, ATC2, ATC3(未分化甲状腺癌)vs PTC1, PTC2, PTC3(乳头状甲状腺癌)
- 总reads数:各样本测序深度约数百万(真实测序深度)
- 零值比例:约20-50%,符合RNA-seq数据特征(大量低表达/未表达基因)
</details>
---
## 第二部分:DESeq2对象创建与质控
### 练习2.1:创建DESeq2对象
```{r}
# TODO: 创建样本信息表并构建DESeq2对象
# 1. 创建colData数据框(样本信息)
# 从sample_info中提取必要信息
colData <- data.frame(
condition = conditions,
batch = sample_info$batch, # 批次信息(C1, C2)
row.names = sample_info$sample # 使用样本名作为行名
)
cat("样本信息表:\n")
print(colData)
# 2. 创建DESeqDataSet对象
# 注意:DESeq2需要原始counts,不要使用标准化后的数据!
dds <- DESeqDataSetFromMatrix(
countData = counts,
colData = colData,
design = ~ condition # 设计公式:比较condition间的差异
)
cat("\nDESeq2对象信息:\n")
print(dds)
cat("\n设计公式:", as.character(design(dds)), "\n")
cat("\n注意:数据中包含批次信息(C1, C2),可用于后续的批次效应分析\n")
```
<details>
<summary>点击查看核心代码</summary>
```r
colData <- data.frame(
condition = conditions,
batch = sample_info$batch,
row.names = sample_info$sample
)
dds <- DESeqDataSetFromMatrix(
countData = counts,
colData = colData,
design = ~ condition
)
```
</details>
::: {.callout-warning}
### ⚠️ 常见错误
**错误**:使用TPM/FPKM等标准化数据创建DESeq2对象
**正确**:DESeq2必须使用原始count数据,标准化在内部自动进行
:::
---
### 练习2.2:过滤低表达基因
**为什么需要过滤?**
- 低表达基因的计数随机性大,可能引入噪音
- 减少计算量,提高统计效能
```{r}
# TODO: 完成低表达基因过滤
# 1. 统计过滤前的基因数
genes_before <- nrow(dds)
cat("过滤前基因数:", genes_before, "\n")
# 2. 过滤条件:至少3个样本中counts >= 10
# 提示:使用 rowSums() 和条件筛选
keep <- rowSums(counts(dds) >= 10) >= 3
# 3. 应用过滤
dds_filtered <- dds[keep, ]
# 4. 统计过滤后的基因数
genes_after <- nrow(dds_filtered)
cat("过滤后基因数:", genes_after, "\n")
cat("过滤比例:", round((1 - genes_after/genes_before) * 100, 1), "%\n")
# 5. 更新dds对象
dds <- dds_filtered
cat("\n过滤后的DESeq2对象:\n")
print(dds)
```
<details>
<summary>点击查看核心代码</summary>
```r
genes_before <- nrow(dds)
cat("过滤前基因数:", genes_before, "\n")
keep <- rowSums(counts(dds) >= 10) >= 3
dds_filtered <- dds[keep, ]
genes_after <- nrow(dds_filtered)
cat("过滤后基因数:", genes_after, "\n")
cat("过滤比例:", round((1 - genes_after/genes_before) * 100, 1), "%\n")
dds <- dds_filtered
```
</details>
---
### 练习2.3:计算Size Factors(测序深度标准化)
**原理**:中位数比值法
```
size factor_j = median_i (counts_ij / geometric_mean_i)
```
假设:大多数基因在不同样本间表达稳定
```{r}
# TODO: 完成size factor计算和分析
# 1. 计算每个样本的总reads数
lib_size <- colSums(counts(dds))
cat("各样本测序深度(总reads数):\n")
print(lib_size)
# 2. 计算size factors(DESeq2自动)
dds <- estimateSizeFactors(dds)
# 3. 提取size factors
size_factors <- sizeFactors(dds)
cat("\nSize Factors:\n")
print(round(size_factors, 3))
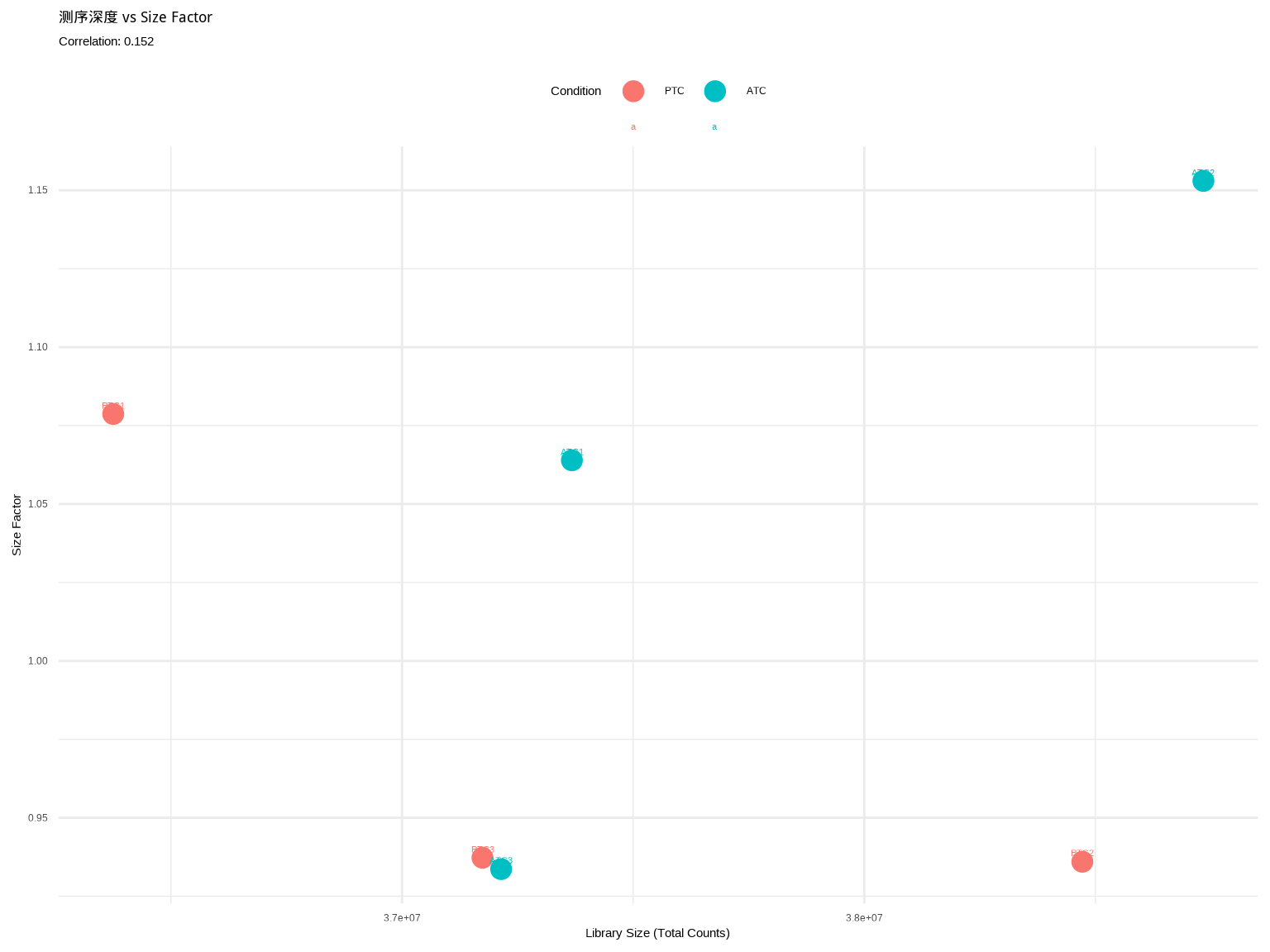
# 4. 分析:size factor与测序深度的关系
correlation <- cor(lib_size, size_factors)
cat("\n测序深度 vs Size Factor 相关性:", round(correlation, 3), "\n")
# 5. 可视化
df_plot <- data.frame(
Sample = colnames(dds),
LibSize = lib_size,
SizeFactor = size_factors,
Condition = colData(dds)$condition
)
ggplot(df_plot, aes(x = LibSize, y = SizeFactor, color = Condition)) +
geom_point(size = 4) +
geom_text(aes(label = Sample), vjust = -0.8, size = 3) +
theme_minimal() +
labs(title = "测序深度 vs Size Factor",
subtitle = paste("Correlation:", round(correlation, 3)),
x = "Library Size (Total Counts)",
y = "Size Factor") +
theme(legend.position = "top")
```
<details>
<summary>点击查看核心代码</summary>
```r
lib_size <- colSums(counts(dds))
dds <- estimateSizeFactors(dds)
size_factors <- sizeFactors(dds)
# 可视化代码同上
```
</details>
::: {.callout-note}
### 💡 关键理解
- Size Factor > 1:该样本测序深度高于中位数,counts会被缩小
- Size Factor < 1:该样本测序深度低于中位数,counts会被放大
- 理想情况下,Size Factor应与测序深度正相关
:::
::: {.callout-important}
### 🔍 深入理解:为什么 Library Size 大但 Size Factor 可能 < 1?
这是学习者常遇到的困惑!关键在于理解 **Size Factor ≠ Library Size 归一化**。
**DESeq2 的中位数比值法原理**:
```
size factor_j = median_i (counts_ij / geometric_mean_i)
```
对于每个基因 i,计算其在所有样本中的几何平均值,然后计算每个样本中该基因与几何平均值的比值,最后取所有基因比值的中位数。
**可能出现 "Library Size 大但 Size Factor < 1" 的原因**:
1. **表达分布差异**:PTC2 虽然总 reads 多,但这些 reads 可能集中在少数高表达基因上,而大多数基因的表达量相对较低。
2. **几何平均效应**:如果一个样本中大多数基因(特别是中等表达基因)的表达量低于几何平均值,即使有少数超高表达基因"贡献"了大量 reads,size factor 仍可能 < 1。
3. **RNA 组成偏好**:某些生物学条件可能导致特定基因大幅上调,"吸收"了大量测序资源,导致其他基因的相对比例下降。
**类比理解**:
想象两个国家的人均收入比较:
- A国:总收入100亿,人口1亿,但90%的财富集中在1%的富人手中
- B国:总收入80亿,人口1亿,财富分配相对均匀
虽然A国总收入更高,但如果看"中位数收入",B国可能更高。Size Factor 就像"中位数收入",不受极端值影响。
**如何验证**:
可以检查 PTC2 样本中基因表达的分布:
- 计算基因表达的 Gini 系数(不平等程度)
- 比较高表达基因占比
- 查看是否存在少数极高表达基因
这是 DESeq2 方法设计的优势——对 RNA 组成偏好具有稳健性!
:::
---
## 第三部分:数据转换与可视化
### 练习3.1:获取标准化和转换后的数据
```{r}
# TODO: 获取不同类型的数据
# 1. 获取normalized counts(size factor校正后)
normalized_counts <- counts(dds, normalized = TRUE)
cat("原始counts示例(前3个基因,前3个样本):\n")
print(head(counts(dds)[, 1:3], 3))
cat("\n标准化counts示例(相同基因和样本):\n")
print(round(head(normalized_counts[, 1:3], 3), 2))
# 2. 使用VST进行数据转换
# VST (Variance Stabilizing Transformation) 使方差趋于稳定
dds_vst <- vst(dds, blind = FALSE) # blind=FALSE使用设计信息
# 3. 提取转换后的矩阵
vst_matrix <- assay(dds_vst)
cat("\nVST转换后数据(前3个基因,前3个样本):\n")
print(round(vst_matrix[1:3, 1:3], 2))
cat("\nVST数据范围:", round(min(vst_matrix), 2), "to", round(max(vst_matrix), 2), "\n")
```
<details>
<summary>点击查看核心代码</summary>
```r
normalized_counts <- counts(dds, normalized = TRUE)
dds_vst <- vst(dds, blind = FALSE)
vst_matrix <- assay(dds_vst)
```
</details>
::: {.callout-note}
### VST vs rlog 选择
- **VST**:适用于大样本(n > 30),计算更快
- **rlog**:适用于小样本(n < 30),效果更稳定
- 本实验使用VST即可
:::
---
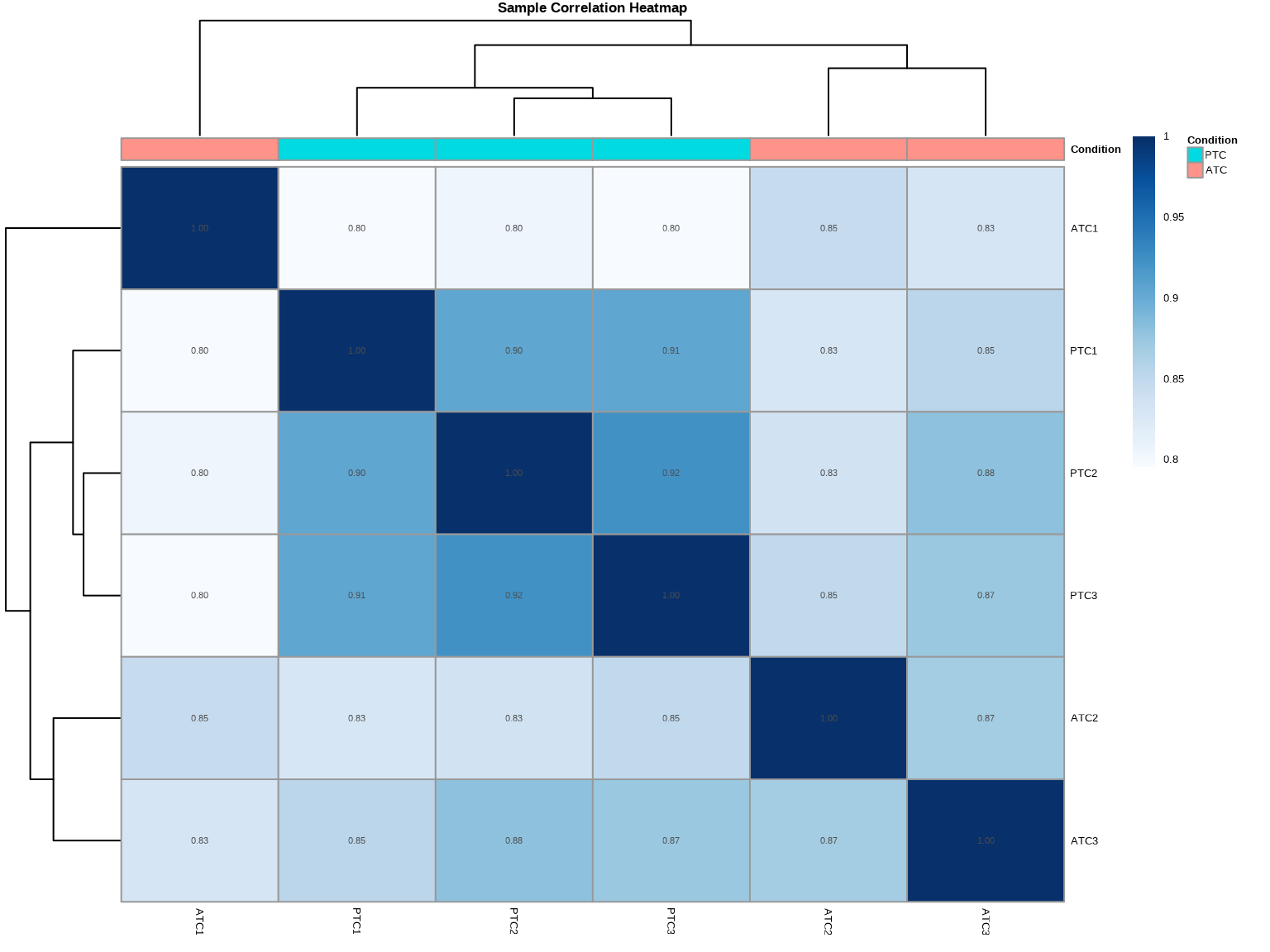
### 练习3.2:样本间相关性分析
```{r}
# TODO: 计算和可视化样本间相关性
# 1. 计算样本间Pearson相关系数矩阵
cor_matrix <- cor(vst_matrix)
cat("样本间相关性矩阵:\n")
print(round(cor_matrix, 3))
# 2. 准备注释数据
annotation_col <- data.frame(
Condition = colData(dds)$condition,
row.names = colnames(dds)
)
# 3. 绘制相关性热图
pheatmap(cor_matrix,
annotation_col = annotation_col,
main = "Sample Correlation Heatmap",
color = colorRampPalette(brewer.pal(9, "Blues"))(100),
display_numbers = TRUE,
fontsize_number = 8)
```
<details>
<summary>点击查看核心代码</summary>
```r
cor_matrix <- cor(vst_matrix)
annotation_col <- data.frame(
Condition = colData(dds)$condition,
row.names = colnames(dds)
)
pheatmap(cor_matrix,
annotation_col = annotation_col,
main = "Sample Correlation Heatmap",
color = colorRampPalette(brewer.pal(9, "Blues"))(100))
```
</details>
::: {.callout-important}
### 质控判断标准
✅ **良好**:组内相关性 > 组间相关性(同条件样本聚类在一起)
❌ **异常**:样本按批次或其他非生物学因素聚类
:::
---
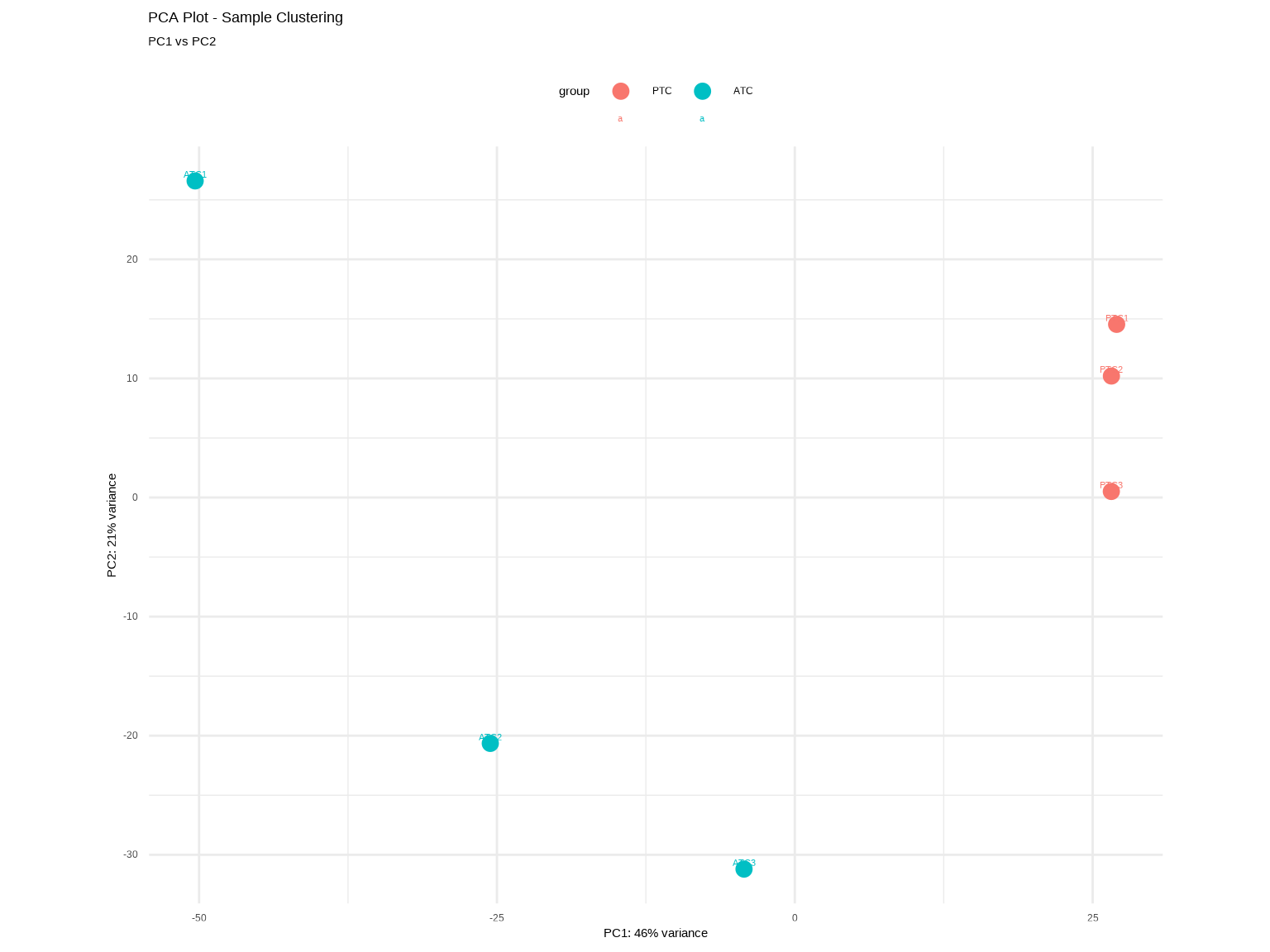
### 练习3.3:主成分分析(PCA)
PCA帮助我们识别样本间的主要变异来源和潜在的批次效应。
```{r}
# TODO: 完成PCA分析
# 1. 使用DESeq2的plotPCA函数
pca_plot <- plotPCA(dds_vst, intgroup = "condition")
# 2. 美化PCA图
pca_plot +
theme_minimal() +
geom_text(aes(label = name), vjust = -0.5, size = 3) +
labs(title = "PCA Plot - Sample Clustering",
subtitle = "PC1 vs PC2") +
theme(legend.position = "top")
# 3. 提取PCA数据并查看方差解释比例
pca_data <- plotPCA(dds_vst, intgroup = "condition", returnData = TRUE)
percentVar <- round(100 * attr(pca_data, "percentVar"))
cat("\nPCA方差解释:\n")
cat("PC1 解释方差:", percentVar[1], "%\n")
cat("PC2 解释方差:", percentVar[2], "%\n")
cat("PC1 + PC2 累计:", sum(percentVar[1:2]), "%\n")
# 4. 解读问题(请回答)
cat("\n=== PCA解读 ===\n")
cat("1. PC1是否主要分离PTC和ATC样本?", "\n")
cat("2. 组内样本是否聚类在一起?", "\n")
cat("3. 是否有明显的异常样本?", "\n")
```
<details>
<summary>点击查看核心代码</summary>
```r
pca_plot <- plotPCA(dds_vst, intgroup = "condition")
pca_plot +
theme_minimal() +
geom_text(aes(label = name), vjust = -0.5, size = 3) +
labs(title = "PCA Plot - Sample Clustering")
pca_data <- plotPCA(dds_vst, intgroup = "condition", returnData = TRUE)
percentVar <- round(100 * attr(pca_data, "percentVar"))
```
</details>
---
## 第四部分:综合质控报告
### 练习4.1:生成质控报告
基于上述分析,完成以下质控报告:
```r
# TODO: 基于分析结果回答以下问题
cat("==============================================\n")
cat(" RNA-seq 质控报告\n")
cat("==============================================\n\n")
# 1. 数据过滤
cat("1. 数据过滤\n")
cat(" - 原始基因数:", genes_before, "\n")
cat(" - 过滤后基因数:", genes_after, "\n")
cat(" - 过滤比例:", round((1 - genes_after/genes_before) * 100, 1), "%\n")
cat(" - 判断: ___ (是否合理? 一般过滤30-50%为正常)\n\n")
# 2. Size Factors
cat("2. Size Factors\n")
cat(" - 范围:", round(min(size_factors), 3), "to", round(max(size_factors), 3), "\n")
cat(" - 与测序深度相关性:", round(correlation, 3), "\n")
cat(" - 判断: ___ (差异是否过大? 一般0.5-2.0为正常)\n\n")
# 3. 样本相关性
group1_cor <- mean(cor_matrix[1:3, 1:3][lower.tri(cor_matrix[1:3, 1:3])])
group2_cor <- mean(cor_matrix[4:6, 4:6][lower.tri(cor_matrix[4:6, 4:6])])
between_cor <- mean(cor_matrix[1:3, 4:6])
cat("3. 样本间相关性\n")
cat(" - PTC组内平均相关性:", round(group1_cor, 3), "\n")
cat(" - ATC组内平均相关性:", round(group2_cor, 3), "\n")
cat(" - 组间平均相关性:", round(between_cor, 3), "\n")
cat(" - 判断: ___ (组内 > 组间? 这是理想情况)\n\n")
# 4. PCA
cat("4. PCA分析\n")
cat(" - PC1解释方差:", percentVar[1], "%\n")
cat(" - PC2解释方差:", percentVar[2], "%\n")
cat(" - 判断: ___ (PC1是否分离两组样本?)\n\n")
# 5. 总体评估
cat("5. 总体质控结论\n")
cat(" - 数据质量: ___ (良好/一般/差)\n")
cat(" - 是否可继续差异分析: ___ (是/否)\n")
cat(" - 注意事项: ___\n\n")
cat("==============================================\n")
```
---
## 思考题
1. **为什么需要过滤低表达基因?**
<details>
<summary>点击查看答案</summary>
低表达基因的计数随机性大,可能引入噪音;统计检验效能不足
</details>
2. **Size Factor与简单CPM标准化的区别?**
<details>
<summary>点击查看答案</summary>
Size Factor使用中位数比值法,对高表达基因稳健;CPM易受极值影响
</details>
3. **如果PCA显示PTC和ATC没有清晰分离,可能是什么原因?**
<details>
<summary>点击查看答案</summary>
处理效应弱、批次效应、样本标签错误或实验失败
</details>
4. **VST转换的目的是什么?**
<details>
<summary>点击查看答案</summary>
使数据方差趋于稳定,适合PCA等需要正态分布假设的分析
</details>
---
## 关键代码速查表
| 操作 | 代码 |
|------|------|
| 创建DESeq2对象 | `DESeqDataSetFromMatrix(countData, colData, design = ~ condition)` |
| 过滤低表达基因 | `dds[rowSums(counts(dds) >= 10) >= 3, ]` |
| 计算Size Factors | `estimateSizeFactors(dds)` |
| 获取normalized counts | `counts(dds, normalized = TRUE)` |
| VST转换 | `vst(dds, blind = FALSE)` |
| PCA | `plotPCA(vst_obj, intgroup = "condition")` |
| 样本相关性 | `cor(assay(vst_obj))` |
---
## 延伸阅读
- [DESeq2 Vignette - Data Transformation](https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#data-transformations)
- [RNA-seq Data Quality Control Best Practices](https://www.bioconductor.org/packages/release/workflows/html/rnaseqGene.html#data-quality)
---
**实验结束,请继续完成实验2:差异表达分析与可视化** 🎉