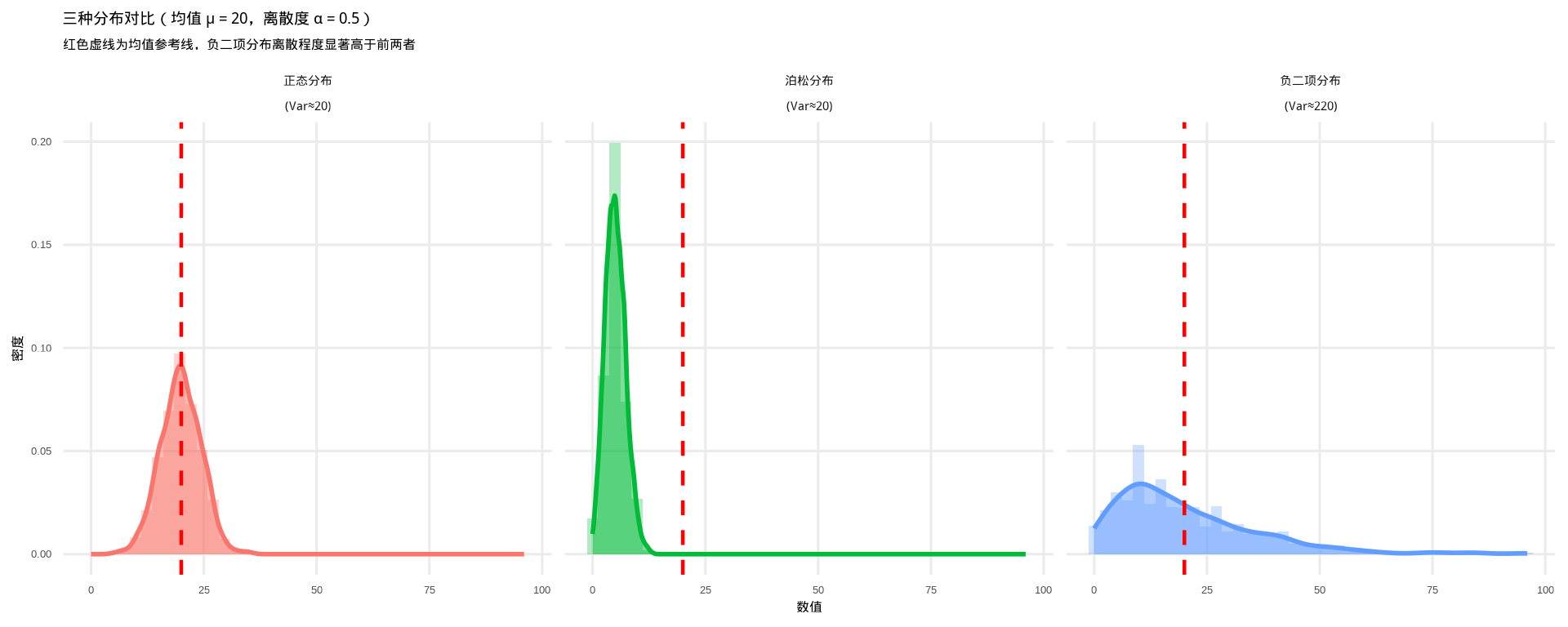
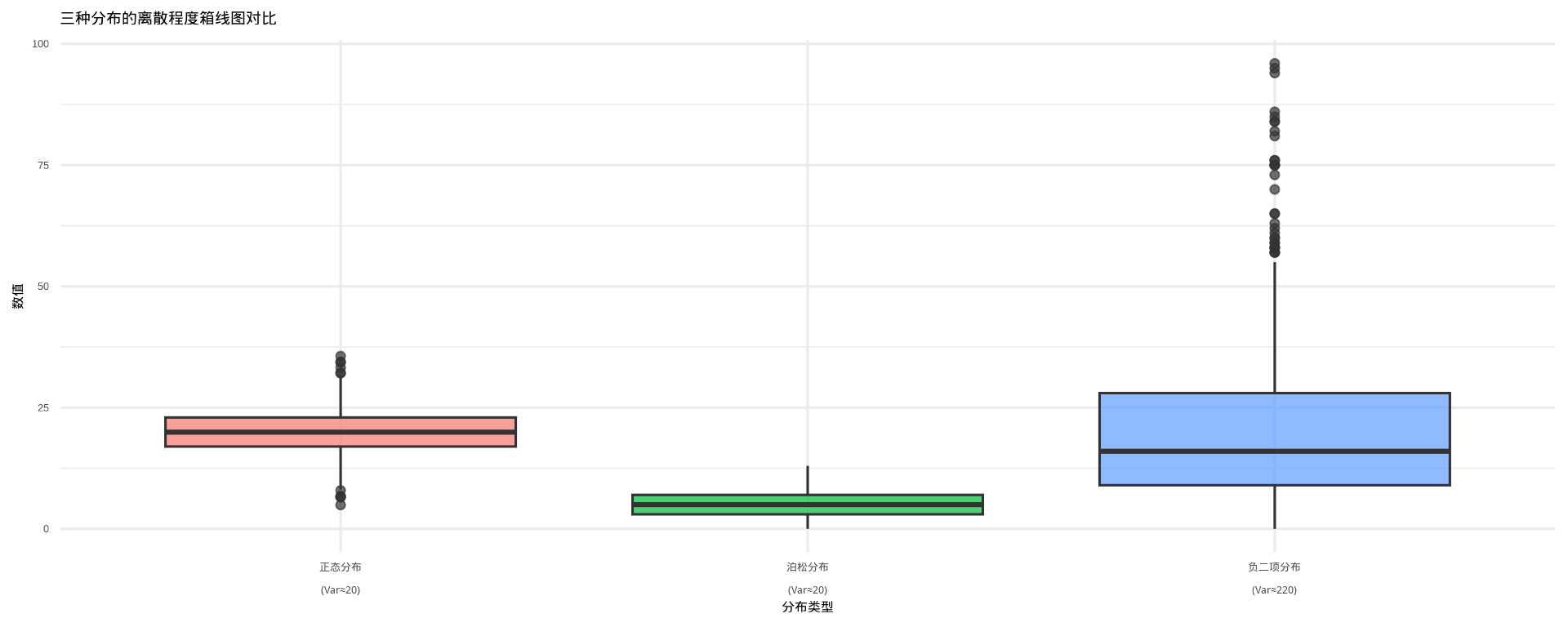
# 1. 设置参数(修正负二项参数,确保方差差异显著)
mu <- 20 # 三种分布的均值统一为20
alpha <- 0.5 # 负二项离散度参数,alpha越大离散程度越高
set.seed(42) # 固定随机种子,结果可复现
# 2. 生成三种分布数据
## 正态分布(t检验假设):方差=均值=20,标准差=sqrt(20)≈4.47
normal_data <- rnorm(1000, mean = mu, sd = sqrt(mu))
## 泊松分布(传统计数模型假设):方差=均值=20/4
poisson_data <- rpois(1000, lambda = mu/4)
## 负二项分布(过离散计数模型):修正参数,确保方差=mu + alpha*mu²=20+0.5*400=220
# R中rnbinom的size参数对应GLM中的theta,和alpha的关系为:size = 1/alpha
size <- 1 / alpha # 此时size=2,方差=20 + 20²/2 = 220
nb_data <- rnbinom(1000, size = size, mu = mu)
# 3. 验证三种分布的统计量(可选,确认参数设置正确)
cat("=== 分布统计量验证 ===\n")=== 分布统计量验证 ===正态分布:均值=19.88,方差=20.10泊松分布:均值=5.05,方差=4.95负二项分布:均值=20.29,方差=246.88# 4. 整理数据为长格式,方便ggplot绘图
df <- data.frame(
value = c(normal_data, poisson_data, nb_data),
distribution = rep(c("正态分布\n(Var≈20)",
"泊松分布\n(Var≈20)",
"负二项分布\n(Var≈220)"), each = 1000)
)
# 5. 可视化对比(修正坐标轴尺度,补充密度曲线)
library(ggplot2)
ggplot(df, aes(x = value, fill = distribution, color = distribution)) +
# 直方图:用密度刻度,方便和密度曲线叠加
geom_histogram(aes(y = after_stat(density)),
bins = 40, alpha = 0.3, position = "identity", color = NA) +
# 叠加核密度曲线,更直观展示分布形态
geom_density(alpha = 0.5, linewidth = 1) +
# 分面:固定坐标轴尺度,强制所有分面使用相同的x/y轴范围,差异更明显
facet_wrap(~ distribution, scales = "fixed") +
# 添加均值参考线
geom_vline(xintercept = mu, linetype = "dashed", color = "red", linewidth = 0.8) +
theme_minimal(base_size = 12) +
labs(x = "数值", y = "密度",
title = "三种分布对比(均值 μ = 20,离散度 α = 0.5)",
subtitle = "红色虚线为均值参考线,负二项分布离散程度显著高于前两者") +
theme(
strip.text = element_text(size = 11, face = "bold"),
legend.position = "none", # 分面后不需要图例
panel.grid.minor = element_blank()
)